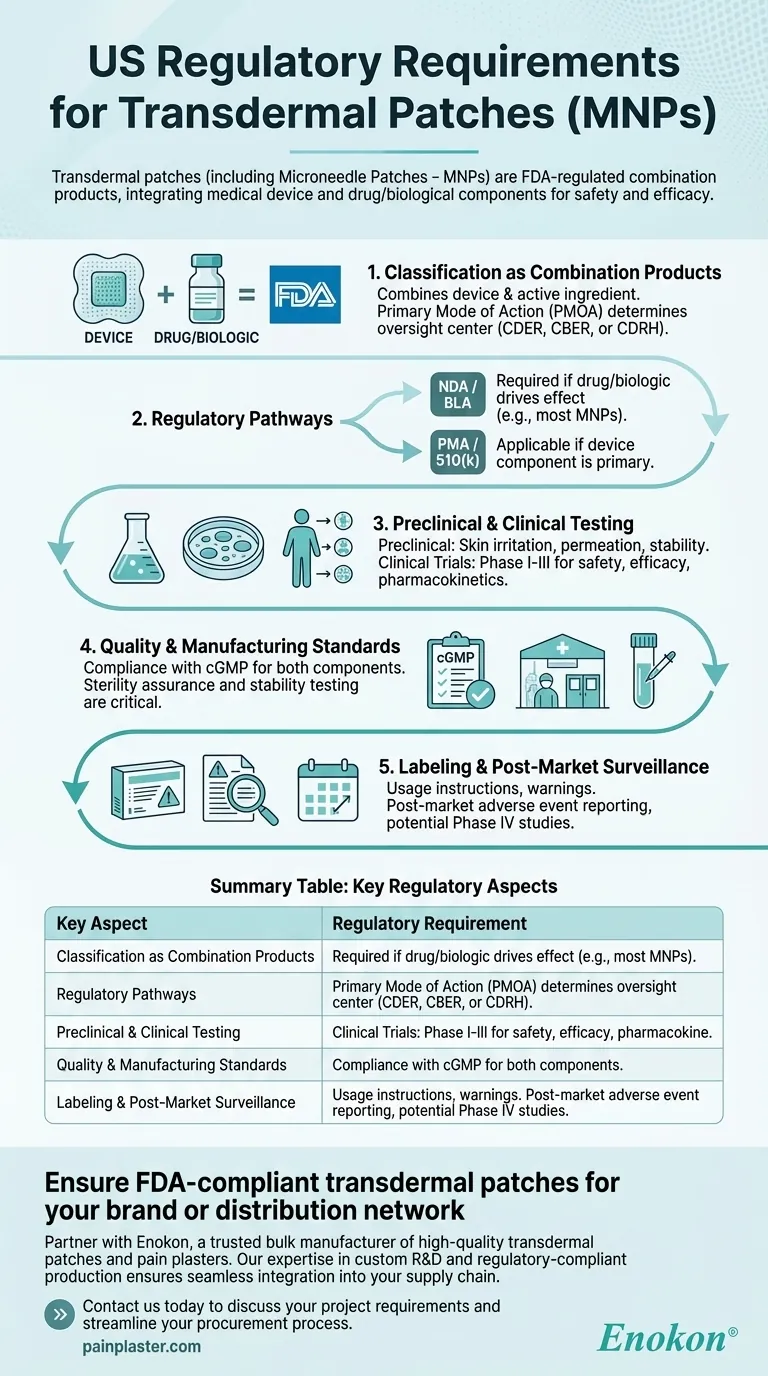
マイクロニードルパッチ(MNP)を含む経皮吸収パッチは、米国ではFDAによりコンビネーション製品として規制されており、安全性と有効性を確保するために厳格な承認プロセスを必要としている。これらの製品は医療機器と薬物・生物学的成分を統合したものであり、主な作用機序に応じて特定の規制経路を遵守する必要がある。承認プロセスには、前臨床試験および臨床試験、品質管理、表示および製造基準の遵守が含まれる。
重要ポイントの説明
-
配合剤としての分類
- FDAは以下のように分類している。 経皮吸収パッチ は、医療機器(例えば、パッチバッキング、マイクロニードル)と薬物または生物学的活性成分を組み合わせたものであるため、コンビネーション製品に分類される。
- 主要作用機序(PMOA)により、製品が医薬品評価研究センター(CDER)、生物製剤評価研究センター(CBER)、または機器・放射線保健センター(CDRH)のいずれで規制されるかが決定される。
-
規制パスウェイ
- 新薬承認申請(NDA)または生物製剤承認申請(BLA):医薬品または生物学的製剤が治療効果をもたらす場合に必要。
- 市販前承認(PMA)または510(k):デバイスのコンポーネントが一次的なものである場合に適用される(例:薬物送達を促進するマイクロニードル)。
- MNPは薬物中心の機能のため、NDA/BLAルートに従うことが多い。
-
前臨床試験と臨床試験
- 前臨床試験:安全性と送達効率を評価するため、皮膚刺激性、浸透性、安定性試験を含む。
- 臨床試験:第I~III相試験では、ヒトにおける薬物動態、有効性、副作用を評価する。
-
品質および製造基準
- 医薬品と医療機器の両方のコンポーネントについて、現行の適正製造基準(cGMP)を遵守します。
- 無菌性保証と安定性試験は、生物製剤を使用するパッチにとって極めて重要である。
-
ラベル表示と市販後調査
- ラベルには、使用方法、警告、保管条件などを記載しなければならない。
- 市販後要件には、有害事象の報告や第IV相試験の可能性が含まれる。
購入者にとっては、これらの要件を理解することで、FDAに準拠したサプライヤーとの連携が確保され、不適合製品のリスクが軽減される。これらの規制が、貴社の調達スケジュールやベンダー選定基準にどのような影響を与えるか検討されたことはありますか?機器と医薬品の監視の相互作用が、最新の経皮吸収型製剤の信頼性を静かに形作っている。
要約表
| 主な側面 | 規制要件 |
|---|---|
| 分類 | FDAによりコンビネーション製品(デバイス+薬剤/生物学的)として規制されている。 |
| 主要作用機序 | CDER(医薬品)、CBER(生物学的製剤)またはCDRH(デバイス)による監視を決定する。 |
| 承認経路 | NDA/BLA(医薬品主導型)またはPMA/510(k)(デバイス主導型)。MNPは通常NDA/BLAに従う。 |
| 試験要件 | 前臨床試験(安全性、透過性)および臨床試験(第Ⅰ相~第Ⅲ相)。 |
| 製造基準 | 医薬品と医療機器の両方のコンポーネントに対するcGMPコンプライアンス、生物製剤の無菌性保証。 |
| 市販後の義務 | 有害事象報告、第IV相試験、およびラベリングコンプライアンス。 |
貴社のブランドまたは販売ネットワークにFDA準拠の経皮吸収パッチを保証します。
パートナー
エノコン
高品質な経皮吸収型テープ製剤のバルクメーカーとして信頼されています。カスタムR&Dおよび規制に準拠した製造における当社の専門知識により、貴社のサプライチェーンへのシームレスな統合を実現します。
お問い合わせ
お客様のプロジェクト要件と調達プロセスの合理化についてご相談ください。
ビジュアルガイド